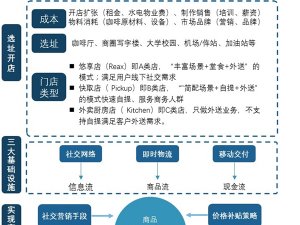
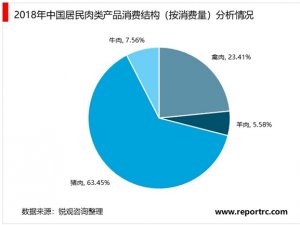
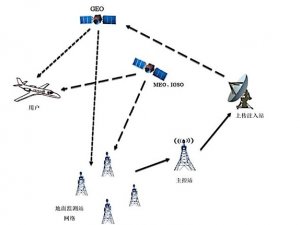
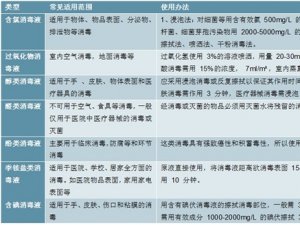
1、我国与美国的器械注册审批差异
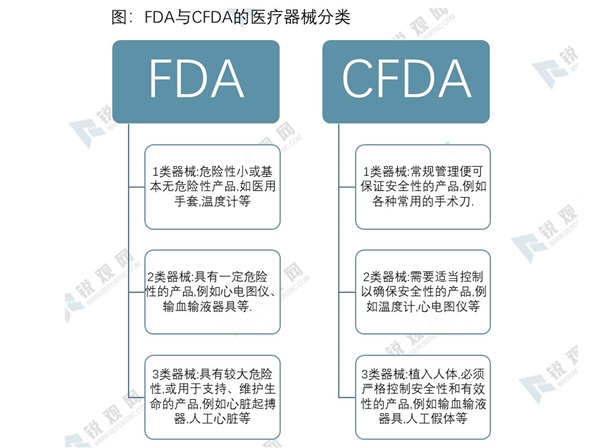
美国同样将医疗器械分为三类。第Ⅰ类为“普通管理”产品——危险性小或基本无危险,例如医用手套、压舌板、手动手术器械、温度计等。第Ⅱ类为“性能标准管理”产品——具有一定危险性的产品,例如心电图仪、超声诊断仪、输血输液器具、呼吸器等。第Ⅲ类为用于支持或维持生命、对保护人类健康起至关重要的作用,或存在导致病痛或伤害的潜在、过度风险的产品,例如人工心脏瓣膜、心脏起搏器、人工晶体、人工血管等。
虽然FDA与CFDA都将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,但具体的器械分类目录有所不同。FDA主要是依据风险等级来进行分类,而CFDA在依据风险等级的同时,也会注重医疗器械的功能、临床使用和管理难度。总的来说,FDA更侧重于产品本身的危险性,而CFDA则更关注产品管理的难易程度。比如输血输液器具,FDA认为器具本身的危险性并不高,因此归于II类,而CFDA认为,输血输液器具会直接侵入人体,若是不严格管理输血输液器具的使用,可能会对人体造成极大危害,例如一器多用有造成血友病,艾滋病,或是误输药剂等风险。造成这一不同的根本原因,是FDA在为器械分类时,默认要求该器械处于一般控制之下,即默认按照基本管理规范来使用器械,而CFDA则考虑到管理不当的风险。
资料来源:锐观咨询整理
2、审评审批流程差异
FDA总部共由生物制品评价研究中心(CBER)、器械和放射产品健康中心(CDRH)、药物评价研究中心(CDER)等部门组成。除血源筛查的医疗器械由生物制品评价研究中心(CBER)负责管理外,其余的医疗器械产品均由器械和放射产品健康中心(CDRH)负责管理。
看上去中美审查流程相差不大,然而在实际申请中会发现差异还是非常明显的。总体来说,FDA宽进严出,CFDA严进宽出。相比FDA,CFDA在审批流程上会非常严格。自2015年7月22日,CFDA发布《关于开展药物临床试验数据自查核查工作的公告》(2015年第117号)以来,曾经存在的大量临床试验数据造假现象急剧减少。CFDA希望中国的医疗行业能够重视临床基础数据的重要性,高标准研发,规范生产。
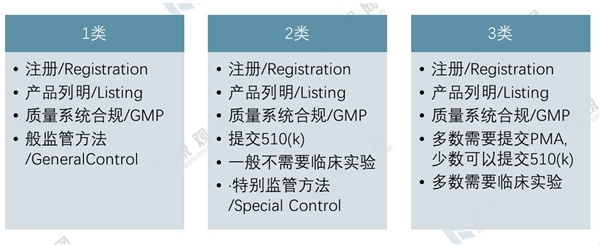
对于任何产品,FDA都要求企业进行企业注册(Registration)和产品列名(Listing)。FDA按风险等级对医疗器械实行分类管理,并由FDA总部对近60%的医疗器械进行上市前通告(510K)或者上市前批准(PMA)的审查。
I类产品为“普通管理(GeneralControls)”产品,是指风险小或无风险的产品,这类产品约占全部美国医疗器械的25%。93%I类和9%II类器械豁免上市前通告程序,只需生产企业确认其产品符合相关规定,并由生产企业向FDA提交保证其产品符合GMP的备案表后,这类产品就能够上市销售。
81%II类器械和小部分III类产品为“普通+特殊管理(General&SpecialControls)”产品,其管理是在“普通管理”的基础上,再通过实施标准管理或特殊管理,以保证质量和安全有效性的产品,这类产品约占全部医疗器械的55%。FDA只对少量的II类产品豁免上市前通告程序,其余大多数产品均要求上市前90天向FDA提出申请进行上市前通告(510K)。上市前通告是上市前通过对拟上市产品与已上市产品在安全性和有效性方面进行比较,在得出实质性等同(SubstantialEquivalence,SE)结论的前提下,进而获得拟上市产品可以合法销售的上市前通告的一条法规路径。
图:FDA对于不同分类医疗器械的审评审批
资料来源:锐观咨询整理
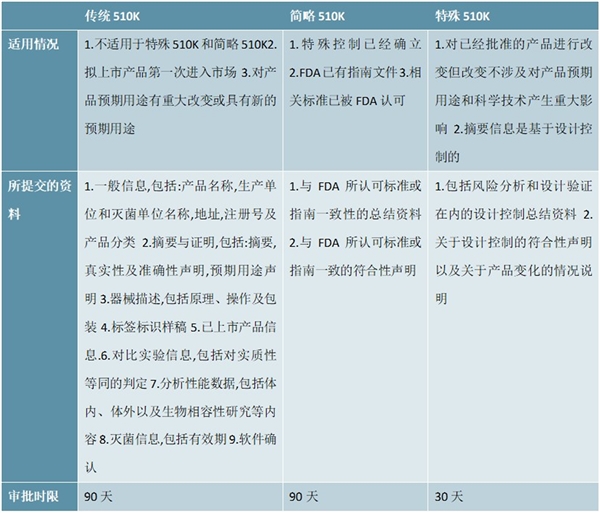
绝大多数Ⅱ类产品都属于需要进行上市前通告(510K)的产品,这类产品是在普通管理的基础上增加一些特殊要求,如:对标识的特殊要求、符合某些性能标准、符合FDA的指南等,以确保其临床使用中的安全性和有效性。这类产品通常要由申请人提交资料证明其与已上市产品实质性等同(SE),经过FDA审查并取得510K确认信后方可上市销售。同时,因其所涉及的具体情况不同,上市前通告又分为传统510K(Traditional510K)、简略510K(Abbreviated510K)和特殊510K(Special510K)三个类型。申请人应根据拟申请上市产品的情况在产品上市90天前向FDA提出不同的510K申请并报送相关资料。
表:三种类型510K申请对照
资料来源:锐观咨询整理
可由第三方(ThirdParty)审核的产品:对于占有60%美国市场份额的II类产品,FDA在麻醉、心血管、临床化学、牙科、耳鼻喉、肠胃、整形外科、常规设备、血液学、免疫学、微生物学、神经学、产科、眼科、病理学、物理治疗学、放射学和毒物学等15类产品范畴中,抽取了部分产品(I类和II类需要完成510(K)申报方能进入美国市场、不属于植入式、支持或维护生命设备、且不需要进行人体临床研究的产品)授权给了11家机构进行第三方审核,从而加快了这类产品完成美国市场准入的进度。当然,对于可由第三方审核的医疗器械,制造商仍然可以向FDA直接申请市场准入审核,而并非必须要经由第三方机构。
绝大多数III类产品为“上市前批准管理(Pre-marketApproval,PMA)”产品,是指具有较高风险或危害性,或是支持或维护生命的产品,这类产品约占全部医疗器械的20%。FDA对此类产品采用上市前批准制度,生产企业在产品上市前必须向FDA提交PMA申请书及相关资料,证明产品质量符合要求,在临床使用中安全有效。FDA在收到PMA申请后45天内通知生产企业是否对此申请立案审查,并在180天(不包括生产企业重新补充资料的时间)内对接受的申请做出是否批准的决定,只有当FDA做出批准申请的决定后,该产品才能上市销售。
3、临床研究管理差异
在美国,有10%-15%的II类产品在申请上市前通告(510K)及全部III类产品在申请上市前批准(PMA)时,都必须提交临床研究资料。对于需要提交临床研究资料的产品,根据其风险程度的不同,又分为具有重大风险的器械(SignificantRiskDevice)和不具有重大风险的器械(Non-SignificantRiskDevice)。
a)具有重大风险的器械:定义为植入人体,用于支持或维持生命,对于诊断、治疗、减轻或处理疾病有重要作用或者防止人体健康受到损害的器械。如果试验发起人判断拟进行临床研究的器械为具有重大风险的器械,那么试验发起人应当按照21CFR812.20的要求向FDA提交一份完整的IDE
(InvestigationalDeviceExemptions)申请的同时,按照21CFR812.25和21CFR812.27的要求向机构审查委员会IRB(InstitutionalReviewBoard)提交临床研究计划和预先研究报告。在经这两个机构审查并获得批准后,临床研究方可开展。
b)具有非重大风险的器械:即是除具有重大风险器械以外的器械,此类器械进
行临床研究不必向FDA提交IDE申请,只需按照21CFR812.2(b)的要求直接向IRB提交简略IDE申请,并提交拟进行临床研究的地点和判断拟进行临床研究的器械不具有重大风险的依据,经审查获得批准后临床研究即可开展。
4、上市后监管
FDA要求生产企业保证产品是在符合GMP要求的条件下生产出来的。在产品上市后,FDA会通过质量体系检查,建立追溯制度,不良事件报告,召回等手段来进行监管。
FDA在监管上特别严格,对产品上市后的持续监控非常看重,它们认为——“虽然上市前临床试验提供了有关设备安全性和有效性的重要信息,但一旦设备投入市场,新安全问题就有可能出现。”这样的后续监控通过几个方面来确保:首先是FDA对美国医械商制造设施的例行检查,以确保其遵循良好的制造规范,并对不符合者予以关闭;另一方面是“问题报告”机制,FDA有几个通路以供制造商、卫生专业人士及消费者反馈设备相关问题:MedWatch—FDA的不良事件报告项目,是报告医疗产品问题及了解最新安全信息的门户,受众可常规订阅MedWatch安全警报;MedSun—医疗产品安全网络,通过在全美招募的350名医疗服务提供者,检测医疗产品的安全性和有效性,报告任何导致严重伤害或死亡的医疗问题,并且每月发布MedSun通讯。

锐观网倡导尊重与保护知识产权。如发现本站文章存在版权问题,烦请联系service@reportrc.com、010-5716921,我们将及时沟通与处理。