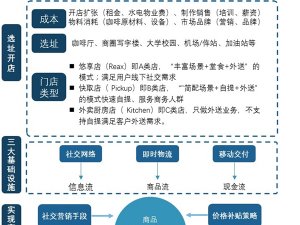
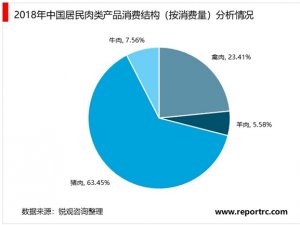
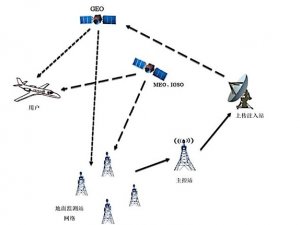
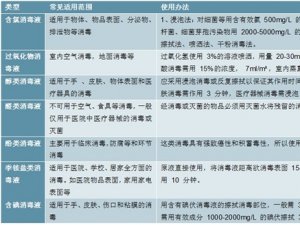
欧盟各成员国的主管当局授权第三方公告机构负责评审和监管。医疗器械可以在欧盟国家上市销售,代表该器械在欧盟境内满足相关指令的基本要求,能合法使用CE标志,并已完成了合格评定程序,能在欧盟成员国内别合法地投放市场。所有医疗器械IIa类及以上的上市前评审由第三方公告机构按照相关的指令进行。I类产品上市前审批不需要公告机构介入,生产企业按照93/42/EEC指令出一个符合性声明就可以合法使用CE标志。另外,含药器械还需要向当地主管当局申请。
产品认证前应先确定产品的分类和认证途径,Ⅱa和Ⅱb类的审批过程基本上是一致的,Ⅲ类相对要更复杂一些,非无菌设备Ⅰ级非测量性器械的流程最简单。然后拟制认证文件,公告机构对企业质量体系和产品技术文件进行审核。初审后公告机构将指出质量体系和技术文件中存在的问题,企业应据此在90天内补充完善质量体系和技术文件。在公告机构审核通过后,对于CE产品证书已经覆盖的产品,可以打上CE标识,CE后面带上机构代码。
体外诊断医疗器械的指令98/79/ECInVitroDiagnosticDirectives(IVDD)
欧洲委员会于1998年10月27日正式通过98/79/EC体外诊断医疗器材指令(InVitroDiagnosticMedicalDevicesDirective,以下简称IVDD指令),并公告于1998年12月7日的第L331号欧盟公报上。根据公报的内容,欧盟各成员国必须于2000
年6月7日之前完成执行本指令所需要的相关法规命令修制定与公告,自2003年12月起,所有在欧盟各成员国销售的体外诊断医疗器材(InVitroDiagnosticMedicalDevices,以下简称IVD)均须依照本指令完成符合性评价程序,贴上CE标示,才能在欧盟上市。
制造商首先要根据体外诊断医疗器材的定义,确定该产品是否为体外诊断医疗器材。下一步就要确定它的分类,根据IVDD指令要求,IVD产品可分成5类:ListA、ListB、自我检测器材(血糖检测除外)、其他类产品、性能评价器材,每一类的符合性评价途径(也就是获得CE认证的途径)各不相同:
1)ListA产品:分险最高,因此控制也最严,需要进行批批检。
a.决定血型ABO、rhesus(C,c,E,e)、anti-Kell的试剂或试剂产品,包括相关之校正物质与对照物质;
b.检测、确认、量化人体样本以标定HIV感染(HIV1,2)、HTLVI,II及B,C,D
型肝炎的试剂及试剂产品,包括相关之校正物质与对照物质。
2)ListB产品:
a.检测抗D与抗K血型的试剂或试剂产品,包括相关校正物质与对照物质;
b.检测异常红细胞抗体的试剂或试剂产品,包括相关校正物质与对照物质;
c.检测与量化人体样本是否有以下的先天感染,如风疹,弓形虫的试剂或试剂产品,包括相关校正物质与对照物质;
d.用以诊断是否有遗传疾病,苯丙酮酸尿症的试剂或试剂产品,包括相关校正物质与对照物质;
e.检测巨细胞病毒、衣原体感染的试剂或试剂产品,包括相关校正物质与对照物质;
f.检测HLA型式DR,A,B的试剂或试剂产品,包括相关校正物质与对照物质;
g.检测肿瘤标记物前列腺特异抗原(PSA)的试剂或试剂产品,包括相关校正物质与对照物质;
h.特定用于评估21三体综合症风险的试剂或试剂产品,包括相关校正物质与对照物质、软件;
i.血糖自我检测的诊断试剂或试剂产品,包括相关校正物质与对照物质。
3)自我检测器材(血糖检测除外):指制造商制定其用途为可被个人在家中使用的器材。
4)其他类产品:凡不属于上述分类的体外诊断医疗器材,可以分到其他。
性能评器材:指在医学分析实验室中使用,用以评估其性能的器材。

锐观网倡导尊重与保护知识产权。如发现本站文章存在版权问题,烦请联系service@reportrc.com、010-5716921,我们将及时沟通与处理。
中国医疗器械行业竞争格局及主要进入壁垒
医疗器械临床试验相关规定梳理
2020眼科医疗器械行业市场市场发展趋势分析,中国器械药品开启
2020医疗器械行业市场发展前景分析,慢病的管理政策出台推动家
2020医疗器械行业市场发展趋势分析,家用医疗器械市场迎来需求
一次性医疗器械行业发展有利因素及不利因素
医疗器械行业发展有利因素及不利因素
医疗器械行业市场发展趋势分析,疫情推动国产设备高端化打开
中国医疗器械相关政策汇总分析
2020医疗器械行业市场发展前景分析,多因素推动行业发展市场前
医疗器械行业市场现状分析,目前国产占比依旧较低多个领域仍
2020医疗器械行业市场发展趋势分析,疫情影响下迎利好出口形势